








Enfermedad de Fabry, una patología genética que afecta el corazón y los riñones
¿Qué es la enfermedad de Fabry y cuáles son sus síntomas?
La Enfermedad de Fabry es una patología genética, la cual es una enfermedad por deposito lisosomal que produce un deterioro progresivo de los órganos, está ligada al X, es multisistémica, y adicionalmente su detección se hace por pruebas genéticas y la evidencia de manifestaciones que van desde las dermatológicas, renales, cardiovasculares, y cerebrovasculares.
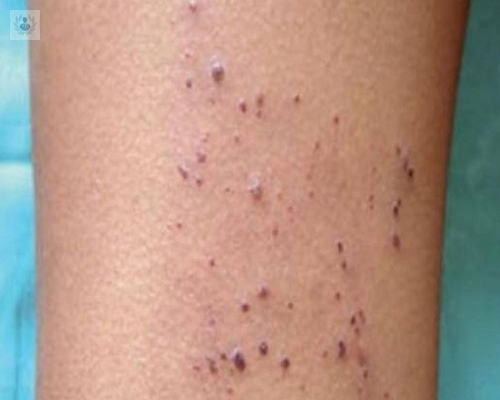
Usualmente en su desarrollo, como es una enfermedad por deposito, comienza desde la infancia por una deficiencia de la alfa-galactosidasa A. Cubre una serie de manifestaciones por órganos y característicamente, en niños se presenta como: alteraciones de la sensibilidad con dolor periférico, disociación de la temperatura corporal, presencia de Angioqueratomas, Proteinuira temprana y Falla Renal de causa no determinada.
A nivel cardiovascular se presentan trastornos inespecíficos de la repolarización en el Electrocardiograma hasta la presencia de Cardiomiopatía Hipertrófica en los pacientes. Usualmente en los pacientes jóvenes desde el punto de vista neurológico, se pueden presentar desde Convulsiones de causa no clara, Accidentes Cerebrovasculares en paciente joven y Sincopes a repetición. Las mujeres afectadas pueden tener síntomas leves y graves. Usualmente, los pacientes pueden experimentar Dolor Crónico con sensación de ardor de la piel y Anhidrosis o Hipohidrosis.
¿Cuáles son los órganos y las funciones que afecta?
En general, la incidencia de la enfermedad es variable con compromiso multiorgánico por el trastorno del metabolismo de los Glicoesfingolipidos, ya sea por déficit o ausencia de actividad de la enzima lisosomal. En general, los órganos afectados son todos. Y en la medida que progresa en el tiempo o se identifica la mutación, puede suceder desde síntomas muy leves a compromiso de órgano severo; por ejemplo: Proteinuria vs Falla Renal Crónica Terminal.
¿Al ser una enfermedad rara, como se puede diagnosticar adecuadamente?
Su diagnostico se establece por la sospecha clínica por la presencia de signos y síntomas que no son identificables para patologías específicas. El deterioro progresivo de los órganos y la valoración genética donde se reporta el déficit o la alteración del gen que codifica la GLA (Xq21.3-q22) que es el que codifica la enzima, lo cual conlleva a la acumulación de una sustancia denominada Gb3 (Globotriaosilceramida) en los lisosomas.
¿En qué consiste el tratamiento?
El tratamiento consiste en la reposición enzimática utilizando la enzima alfa-galactosidasa A, en infusión endovenosa cada 2 semanas o la utilización de chaperonas farmacológicas que permitan mejorar los niveles enzimáticos y que evitan el deterioro progresivo de los órganos por depósitos Gb3.
¿Cuál es la esperanza de llevar una vida normal con esta enfermedad?
En la evolución de la enfermedad se pueden presentar varios escenarios. Desde la aparición temprana de la enfermedad e identificación temprana lo cual mejora el pronóstico de los pacientes. En estudios clínicos se ha determinado que la sobre vida de los pacientes que no se identifican tempranamente o tienen compromiso severo de órganos disminuyen su expectativa de vida por complicaciones cardiovasculares y renales en cerca de 20 años.
Es importante su identificación, dado que tiene penetrancia ligada al X y debe hacerse siempre consejería genética para la identificación de portadores familiares y manejo temprano de la enfermedad por parte de un Especialista.